 Image credit: Unsplash
Image credit: Unsplash
Abstract
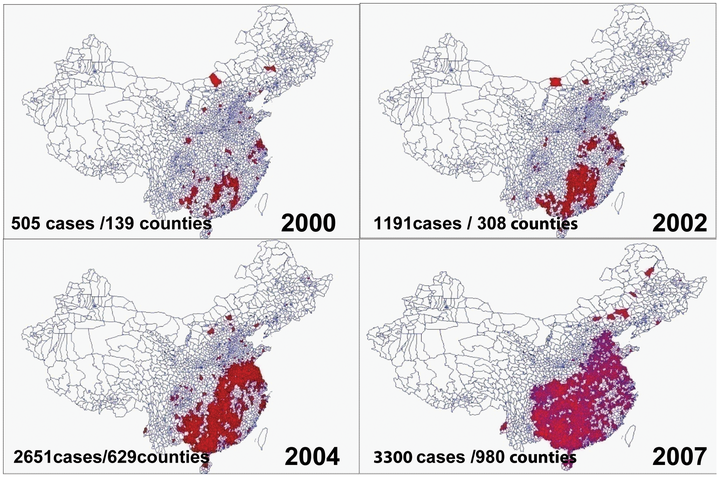
Recent years have seen a rapid increase in the number of rabies cases in China and an expansion in the geographic distribution of the virus. In spite of the seriousness of the outbreak and increasing number of fatalities, little is known about the phylogeography of the disease in China. In this study, we report an analysis of a set of Nucleocapsid sequences consisting of samples collected through the trial Chinese National Surveillance System as well as publicly available sequences. This sequence set represents the most comprehensive dataset from China to date, comprising 210 sequences (including 57 new samples) from 15 provinces and covering all epidemic regions. Using this dataset we investigated genetic diversity, patterns of distribution, and evolutionary history. Our analysis indicates that the rabies virus in China is primarily defined by two clades that exhibit distinct population subdivision and translocation patterns and that contributed to the epidemic in different ways. The younger clade originated around 1992 and has properties that closely match the observed spread of the recent epidemic. The older clade originated around 1960 and has a dispersion pattern that suggests it represents a strain associated with a previous outbreak that remained at low levels throughout the country and reemerged in the current epidemic. Our findings provide new insight into factors associated with the recent epidemic and are relevant to determining an effective policy for controlling the virus.