MiRPara: a SVM-based software tool for prediction of most probable microRNA coding regions in genome scale sequences
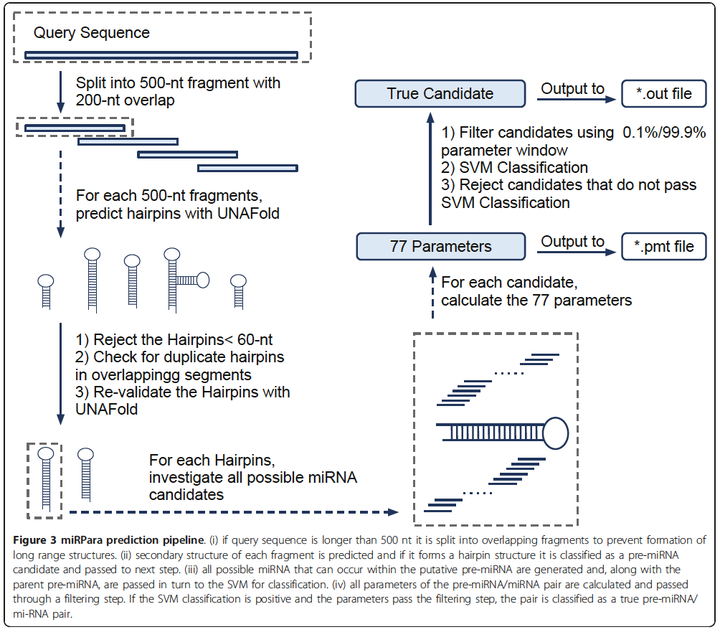 Image credit: Unsplash
Image credit: Unsplash
Abstract
MicroRNAs are a family of ~22 nt small RNAs that can regulate gene expression at the post-transcriptional level. Identification of these molecules and their targets can aid understanding of regulatory processes. Recently, HTS has become a common identification method but there are two major limitations associated with the technique. Firstly, the method has low efficiency, with typically less than 1 in 10,000 sequences representing miRNA reads and secondly the method preferentially targets highly expressed miRNAs. If sequences are available, computational methods can provide a screening step to investigate the value of an HTS study and aid interpretation of results. However, current methods can only predict miRNAs for short fragments and have usually been trained against small datasets which don’t always reflect the diversity of these molecules. We have developed a software tool, miRPara, that predicts most probable mature miRNA coding regions from genome scale sequences in a species specific manner. We classified sequences from miRBase into animal, plant and overall categories and used a support vector machine to train three models based on an initial set of 77 parameters related to the physical properties of the pre-miRNA and its miRNAs. By applying parameter filtering we found a subset of ~25 parameters produced higher prediction ability compared to the full set. Our software achieves an accuracy of up to 80% against experimentally verified mature miRNAs, making it one of the most accurate methods available. miRPara is an effective tool for locating miRNAs coding regions in genome sequences and can be used as a screening step prior to HTS experiments.