miRAW: A deep learning-based approach to predict microRNA targets by analyzing whole microRNA transcripts
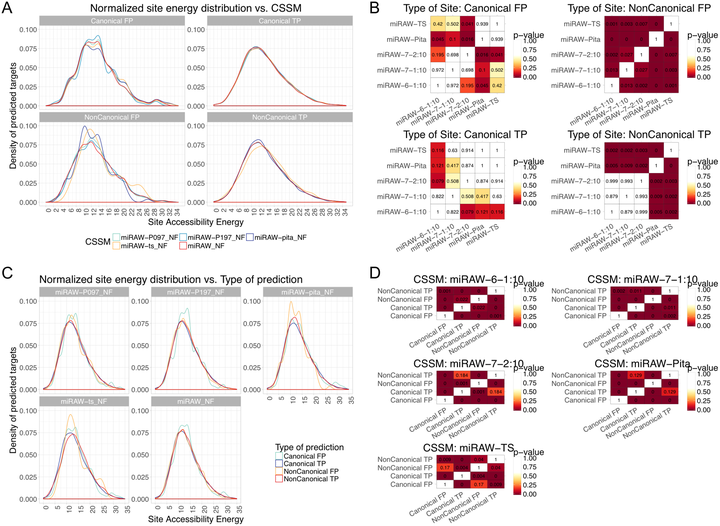 Image credit: Unsplash
Image credit: Unsplash
Abstract
MicroRNAs (miRNAs) are small non-coding RNAs that regulate gene expression by binding to partially complementary regions within the 3’UTR of their target genes. Computational methods play an important role in target prediction and assume that the miRNA “seed region” (nt 2 to 8) is required for functional targeting, but typically only identify ∼80% of known bindings. Recent studies have highlighted a role for the entire miRNA, suggesting that a more flexible methodology is needed. We present a novel approach for miRNA target prediction based on Deep Learning (DL) which, rather than incorporating any knowledge (such as seed regions), investigates the entire miRNA and 3’TR mRNA nucleotides to learn a uninhibited set of feature descriptors related to the targeting process. We collected more than 150,000 experimentally validated homo sapiens miRNA:gene targets and cross referenced them with different CLIP-Seq, CLASH and iPAR-CLIP datasets to obtain ∼20,000 validated miRNA:gene exact target sites. Using this data, we implemented and trained a deep neural network—composed of autoencoders and a feed-forward network—able to automatically learn features describing miRNA-mRNA interactions and assess functionality. Predictions were then refined using information such as site location or site accessibility energy. In a comparison using independent datasets, our DL approach consistently outperformed existing prediction methods, recognizing the seed region as a common feature in the targeting process, but also identifying the role of pairings outside this region. Thermodynamic analysis also suggests that site accessibility plays a role in targeting but that it cannot be used as a sole indicator for functionality.